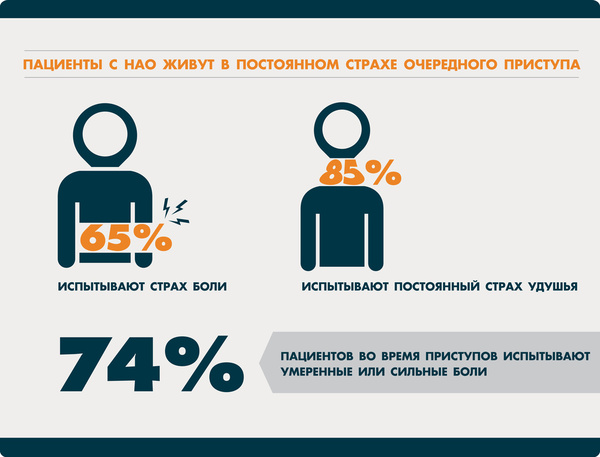
Наследственный ангионевротичекий отёк - редкое, жизнеугрожающее заболевание, которое относится к группе первичных иммунодефицитов. Причина - недостаточность общего уровня или снижение функциональной активности С1-ингибитора системы комплемента. Жизнь таких больных становится кошмаром: они никогда не знают, где и когда начнется отек. Пациенты нередко испытывают страх очередного приступа, для них характерны чувство одиночества, ощущение безысходности и бесконечные проблемы на работе, в учебе и быту.


Наследственный ангионевротический отек (НАО) — хроническое заболевание, относящееся к группе первичных иммунодефицитов с аутосомно-доминантным наследованием и неполной пенетрантностью, связанное с качественным или количественным генетически детерминированным дефектом генов, кодирующих синтез ингибитора эстеразы компонента комплемента C1 (С1inh), которое проявляется в виде рецидивирующих отеков (О.) кожи и слизистых оболочек дыхательных путей, желудочно-кишечного (ЖКТ) и урогенитального трактов [2, 3, 5, 10–14]. Первые упоминания о подобных отёках были сделаны Гиппократом в IV в. до н. э.
Первичный дефицит компонентов комплемента встречается редко, так как для манифестации необходимо гомозиготное состояние по аутосомным аллелям. Исключение — делеция гена С1inh при типе 1, точечная мутация при типе 2 (11р11.2-q13). Больные НАО являются гетерозиготами, т. е. имеют один нормальный и один измененный ген, отвечающий за синтез и функционирование С1inh. Частота мутации гена составляет в среднем 1/100 000 [2, 3, 10, 13]. Несмотря на редкое распространение НАО, в некоторых странах созданы национальные общества и регистры, например, во Франции описано 300 случаев [11].
В настоящее время известны 3 различных дефекта [2, 3, 5, 10–12], которые клинически неразличимы:первый — в 80–92% случаев количественный дефицит С1inh при определении иммунологическими и энзиматическими методами — НАО I типа;второй — в 15% случаев структурный дефект со снижением функциональной активности при нормальном или повышенном уровне С1inh — НАО II типа;третий — в 1–5% случаев структурно измененный С1inh (с возможным ↑ концентрации в 3-4 раза); образуются агрегаты с глобулинами (или альбумином) сыворотки крови или активность С1inh блокирована аутоантителами — НАО III типа. Основной механизм ангиоэдемы при НАО связан с эффектами медиаторов — брадикинина, гистамина, производных арахидоновой кислоты, цитокинов.Критерии диагноза НАО
Положительный генетический (семейный) анамнез в 70–80% случаев (на протяжении 3-4 и даже более поколений).Особенности О.: бледный, очень плотный, имеющий четкую границу со здоровой кожей, захватывающий от 3-4 см в диаметре до больших участков; без гиперемии («холодные О.»), с чувством «напряжения», «распирания тканей». Характерны болезненные О. лица, туловища, конечностей, нередко одной и той же локализации («цикличные О.»). При надавливании на О. ямки не остается.Четкая связь О. кожи и/или слизистых оболочек, абдоминалгий (A.) (табл. 2) с триггерами: механической травмой, интенсивность которой может быть самой разной — от легкого сдавления одеждой или легкого ушиба и до перелома кости (в том числе спортивные и производственные травмы); возникновение О. после экстракции зуба, хирургических операций, диагностических манипуляций инвазивного характера.Возникновение О. кожи и/или слизистых оболочек и А. при интенсивной физической нагрузке, охлаждении, психоэмоциональной перегрузке, на фоне инфекционных заболеваний, менструаций (часто дебют в подростковом возрасте), приеме пероральных контрацептивов, во время беременности.Локализация О.: часто в области дистальных отделов конечностей, верхних дыхательных путей (более 20% случаев) и ЖКТ (более 40% случаев).Сроки развития и динамика изменений О., течение заболевания: О. формируется на протяжении нескольких часов (от 1 до 36 ч), регрессия (самоограничение; после лечения) в течение 10–72 ч (максимально до 7–14 дней). Характерны ремиссии от 7–10 дней до 12 мес, возможны непрерывные атаки, а также латентное (субклиническое) течение.Развитие кольцевидной эритемы (erithema annulare) более чем в 50% случаев.Крапивница, местная гиперемия и зуд не характерны.Абдоминалгии в сочетании с ангиоэдемой или изолированно при атипичном течении.Качественный или количественный дефицит С1inh в момент приступа О. и/или А.Снижение С4-, С2-, C1-компонентов комплемента в периферической крови. При снижении С8, С9 возможно более тяжелое течение.Периферическая эозинофилия, ↑ общего IgE, положительные скарификационные (или прик-) аллергопробы с бытовыми, эпидермальными, пыльцевыми и пищевыми аллергенами не выявляются (возможно редкое сочетание атопии и НАО).Положительный выраженный эффект при в/в введении очищенного С1inh, нативной плазмы, эпсилон-аминокапроновой кислоты (Э-АКК), даназола, станазола, метилтестостерона.Неэффективность антигистаминных препаратов, глюкокортикостероидов (эффект слабый или отсутствует), норадреналина, антибиотиков, противопаразитарных препаратов, ферментов, вегетопротекторов.Особенности клинической картины
Первые признаки НАО могут возникнуть уже в возрасте нескольких месяцев, но чаще после 1-2 лет жизни. У большинства больных НАО дебют заболевания возникает до 20 лет (60%), гораздо реже — в среднем и даже пожилом возрасте [2, 3, 10, 13]. В пубертатном периоде течение заболевания может утяжелиться в связи с гормональной перестройкой. Больные неоднократно наблюдаются у различных специалистов и не сразу обращаются к аллергологу-иммунологу.
Отсутствие в семейном анамнезе сведений о НАО не исключает возможности постановки подобного диагноза [3, 10, 11, 16]. Клинические проявления у пациентов характеризуются рецидивирующими О. различной локализации: кожи лица (губы, периорбитальная область), шеи, туловища, конечностей, слизистых оболочек верхних отделов дыхательных путей, в том числе гортани, желудочно-кишечного (приступообразные боли в животе) и урогенитального трактов. О. может распространяться на верхние дыхательные пути с вовлечением пищевода, гортани и проявляется дисфагией, дисфонией, симптоматикой обструкции дыхательных путей, напоминающей в ряде случаев бронхиальную астму, и прогрессировать вплоть до асфиксии. Смертность при НАО составляет 20–30% [12]. Больным с О. без симптомов крапивницы и кожного зуда следует уделять особое внимание, так как у них может быть АНО с синдромом недостаточности С1inh, носящий наследственный характер.
Интервалы между обострениями у каждого больного индивидуальны: у некоторых больных О. возникают только после значительной травмы, у других обострения проявляются через каждые 9– 14 дней, вне зависимости от внешних воздействий, на протяжении многих лет [2, 3]. Нередко у больных наблюдается аура в виде слабости, разбитости, мраморность кожи, обильных бледных высыпаний типа кольцевидной эритемы, сохраняющейся во время О., не сопровождающиеся зудом, жжением и ↑ t°. О. может возникать на любом участке кожи или слизистых оболочек.
У более половины больных наблюдаются выраженные А. (вызваны О. различных участков слизистой оболочки ЖКТ). Кожные симптомы, как правило, остаются или усиливаются во время О. или А. Развитию приступов A. при НАО часто предшествуют ощущения спазмов в околопупочной области, слабость, тошнота, спастические боли в эпигастрии. Абдоминалгия нередко сопровождается рвотой и жидким стулом, характерна болезненность живота при пальпации. Клинически болевой синдром проявляется в виде разлитой боли в брюшной полости, кишечных колик, возможна тонкокишечная непроходимость. У ряда пациентов наблюдается первичный генез рецидивирующей тонкокишечной непроходимости как forme fruste (приостановление в развитии или скрытое течение заболевания), и только исследование концентрации С4-фракции комплемента позволяет правильно поставить диагноз [7]. Некоторые пациенты предъявляют жалобы на ощущения «отеков внутренних органов».
Боли в животе бывают настолько интенсивными, что их иногда называют «брюшная мигрень», а сопровождающие болевой симптом общие явления (тахикардия, колебания АД, головокружение, головная боль и др.) — «вегетативная буря». Из-за А. больные часто подвергаются лапаротомии; при операции выявляют ограниченный О. кишечника, признаки асептического воспаления.
В более редких случаях при локализации О. на лице могут вовлекаться менингеальные оболочки с проявлением менингеальных симптомов (ригидность затылочных мышц, резкая цефалгия, рвота, иногда судороги), при поражении лабиринтных систем развивается синдром Меньера, что выражается головокружением, тошнотой, рвотой. Все эти симптомы могут встречаться как одновременно, так и по отдельности. При атипичном течении О. могут отсутствовать, также возможны изолированные А., характерны полиартралгии, снижение С4-фракции комплемента. В очень редких случаях описаны: эпилептический приступ, уртикарии, кожная пурпура, феномен Рейно [11]. При лабораторном обследовании больных НАО обнаруживается: С1inh обычно не более 20–30% от нормального (или отсутствие даже в период полной ремиссии), концентрации С2- и С4-компонентов — не более 30–40% от нормальных, уровни С1 и С3 в плазме чаще нормальные. В результате нарушения ингибирования активности С1 постоянно происходит активация комплемента.
Дифференциальная диагностика
Для того чтобы идентифицировать НАО, необходимо провести дифференциальную диагностику генетического, отечного и абдоминального синдромов.
Наследственный вибрационный АНО — самостоятельное заболевание.О. Квинке, крапивница любого происхождения. У 49% больных сочетание крапивницы и О. Квинке, у 40% — только крапивница (в том числе семейная наследственная холодовая крапивница), у 11% — изолированный О. Квинке [10, 17]. Высыпания по типу кольцевидной эритемы возможны не только при НАО, но и при аутоиммунной крапивнице.Описаны приобретенные дефициты С1inh. В этом случае речь идет о «псевдонаследственном ангионевротическом отеке» (ПНАО). Необходимо помнить, что заболевание, проявляющееся характерным клиническим синдромом НАО, не всегда носит наследственный характер. Возможен как приобретенный, так и врожденный характер АНО, связанного с дефицитом С1inh, что может являться следствием случайной мутации (табл. 2, 3).В 80-х гг. J. T. Chiu и соавт. описали редкие случаи приобретенной гипокомплементемии и приступов АНО со снижением содержания С1inh в плазме, что наблюдается при активации и потреблении комплемента при инфекционных заболеваниях и обострении болезней, обусловленных циркуляцией «иммунных комплексов» (ЦИК). Известно редкое явление — персистирующий приобретенный дефицит С1inh в сочетании с ↓ содержания С1, С4 и С2 у больных В-клеточными лимфомами [2, 3, 5, 13, 14; табл. 1, 3].
По сравнению с врожденной формой, приобретенный АНО (ПАНО) с дефицитом С1inh, встречается реже, при этом у других членов семьи больного не обнаруживаются аномалии уровня С1inh. Это комплемент-зависимый ПАНО связан с ускорением метаболизма С1inh в 2-3 раза.
Кроме ↓ уровня С4 и С1inh для больных с приобретенной формой заболевания характерно ↓ С1 и С1q, что помогает проводить дифференциальную диагностику. ПАНО можно отличить от НАО по отсутствию дефектности системы комплемента у здоровых родственников и ↓ содержания С1-компонента, при наследственной форме обычно содержащегося в нормальном количестве (табл. 1).K. E. Binkley и A. Davis в 2000 г. отметили случаи эстроген-зависимой формы НАО, которая наблюдается у женщин. Клинически она не отличается от генетически детерминированного дефицита С1inh (табл. 1). Особенностью этой формы является развитие АНО во время беременности (через 2-3 нед) или через 1-2 нед на фоне приема экзогенных эстрогенов (контрацептивы, гормональная заместительная терапия). Предполагается аутосомно-доминантный тип наследования заболевания, при этом патогенез изучен пока недостаточно; абсолютного или относительного дефицита С1inh у больных до настоящего времени не выявлено.Больным с О. следует проводить тщательное клиническое обследование, так как отечный синдром встречается не только при НАО, ПАНО, но и вследствие хронических заболеваний почек, сердечно-сосудистой системы, эндокринных органов и при других патологиях.Дифференциальный диагноз проводят с редким идиопатическим синдромом Россолимо-Мелькерссона-Розенталя (E. G. Melkersson, 1928; C. Rosenthal, 1931), характеризующимся следующими признаками: 1) рецидивирующие постоянные параличи или неврит лицевого нерва (VII) (Bell, 1836) с чередованием стороны поражения; 2) рецидивирующий О. лица, особенно в области верхнего века и губ; 3) макроглоссия/макроглоссит, макрохейлия; 4) в 30–60% случаев складчатость поверхности языка (lingua plicata). Большинство авторов считают, что наличие 2 из 4 вышеперечисленных признаков позволяет диагностировать данный синдром, при этом обязательно присутствует О. лица. Чаще сочетаются О. губы и неврит лицевого нерва. Симптоматика возникает внезапно, после нескольких частых рецидивов, как правило, наступает длительная ремиссия. При этом исчерченный язык нередко наблюдается и у практически здоровых лиц. Губа приобретает большие размеры, тестоватую (реже плотно-эластическую) консистенцию, красная кайма как бы вывернута. В результате неврита — парез мимических мышц, асимметрия носогубных складок. В период рецидивов описаны периодическое расстройство глотания («глоточные болевые кризы»), слезотечение, пароксизмальные цефалгии, парестезии пальцев. Синдром Россолимо-Мелькерссона-Розенталя может сочетаться с ревматической лихорадкой. При биопсии слизистой оболочки щеки у больных обнаруживаются неказеозные гранулемы. Возможны такие сопутствующие проявления, как ретробульбарный неврит, помутнение роговицы, сухой кератоконъюнктивит.Дифференциальный диагноз также проводят с гранулематозным хейлитом Мишера (G. Miescher, 1945), характеризующимся макрохейлией — стойким воспалительным отеком губ (чаще нижней), длящимся месяцы и годы. Существует мнение, что хейлит Мишера одно из проявлений синдрома Мелькерссона-Розенталя. В 1898 г. Мейж описал ограниченный невоспалительного характера лимфостаз на лице, повторно рецидивирующий и имеющий исходом стойкое утолщение тканей. Причиной этого гистологически является значительное увеличение лимфатических щелей и развитие соединительной ткани в области жировой клетчатки лица. В отличие от синдрома Мелькерссона-Розенталя, не обнаруживают одонто-, рино-, фарингогенных и других очагов хронической инфекции, отсутствуют также пиодермические явления и другие инфекционные заболевания. После каждого рецидива развивается плотный О. различных частей лица.Этиопатогенетическая структура отечного синдрома чрезвычайно вариабельна и имеет полиморфный генез: гипоонкотический, мембраногенный, эндокринный, венозный, лимфогенный О., также отмечены медикаментозные и идиопатические О. [7, 10, с дополнениями].
Распространенные и редкие причины А. могут быть связаны с патологией ЖКТ, гепатобиллиарной системы и поджелудочной железы, системы органов дыхания, сердечно-сосудистой системы, нервной системы, опорно-двигательного аппарата, мочевыделительной системы, половых органов, эндокринного и метаболического генеза, при гематологических заболеваниях и иммунодефицитных состояниях, а также при распространенных инфекционных и паразитарных заболеваниях [12, 14, 16, 18, с дополнениями].
Описанный ниже клинический случай пациентки с направительным диагнозом периодической болезни (ПБ) заставил нас вспомнить об этом заболевании.ПБ, или семейная средиземноморская лихорадка (доброкачественный (семейный) пароксизмальный перитонит, возвратный полисерозит; перемежающаяся шестидневная лихорадка, ложный «острый живот», синдром Джэйнуэя-Мозенталя, болезнь Реймана, синдром Сигала-Коттена-Маму). До недавнего времени ПБ считалась редкой генетически обусловленной патологией с аутосомно-рецессивным (не исключается аутосомно-доминантный) типом наследования и полной пенетрантностью гена [1, 4, 6, 8, 9, 15, 18, с дополнениями].
Болезнь начинается чаще в детстве и встречается у людей из этнических групп Восточного Средиземноморья (дебют первого приступа ПБ, как правило, наблюдают до 30 лет и в 1,5-2 раза чаще у мужчин с астенической конституцией) — армян, евреев (чаще сефардов), арабов, турков, реже у лиц других национальностей. Средний возраст, в котором дебютируют первые признаки болезни, — 9 лет с колебаниями от 2 мес до 60 лет. Средняя продолжительность болезни — 16,8 лет. Ежегодно отмечается тенденция роста заболеваемости и расширение географии распространения ПБ, причем не только у представителей народов средиземноморского бассейна, но и среди коренного населения: у японцев, русских, болгар, итальянцев. Эндемическим районом считается Армения [4, 5, с дополнениями]. Частота семейных случаев ПБ резко варьирует от 6,8 до 60%. По данным А. А. Айвазяна (n=1036) [1], частота семейных форм составляет 27,5%. Вероятность передачи заболевания от матери к дочери — 2,2%, а от отца к сыну — 28%.
Провоцирующими пароксизмы факторами обычно являются: эмоциональная перегрузка, переутомление, охлаждение, интеркуррентные заболевания, различные пищевые продукты, оперативные вмешательства, перемена климата, обострение язвенной болезни и т. д. Для ПБ характерно обострение с началом менструации, ремиссия болезни во время беременности и после назначения больным прогестерона. Продромальными симптомами (81%) при ПБ являются: слабость, недомогание, зевота, чувство внутренней тревоги, снижение аппетита, раздражительность, ломота в теле, бледность кожных покровов или крапивница, акроцианоз, парестезии, ощущение жжения в животе, полидипсия, похолодание конечностей, озноб и повышение температуры тела, которые обычно наблюдаются за 1-2 ч до развития основного синдрома [4]. По основным клиническим проявлениям выделяют: 1) абдоминальную, 2) торакальную, 3) суставную, 4) лихорадочную и 5) смешанную формы ПБ. Характерны стереотипные периодические пароксизмы высокой t до 38–40°С в 100%, А. в 81,7–98% и/или торакалгий (плевриты и перикардиты) в 33–66%, артралгий и/или артритов в 50–77%, рожеподобной сыпи (эритема) в 46%, лимфаденопатии в 1–6% случаев. Патогномоничные симптомы ПБ длятся, как правило, от нескольких часов до 3 сут, проходят самостоятельно. Основу клинической картины асептических перитонитов составляет парез ЖКТ. На высоте приступа интенсивные А. сопровождаются тошнотой, рвотой, вздутием живота, задержкой отхождения газов. При объективном исследовании отмечаются: гиперемия лица, тахикардия, вздутие, напряжение, усиление кишечных шумов и болезненность живота, симптомы раздражения брюшины, при этом живот не участвует в акте дыхания. Часто определяется гепатомегалия, реже — спленомегалия.
При лабораторном исследовании в этот период определяются острофазные воспалительные изменения периферической крови. Лапароскопия считается основным диагностическим тестом, особенно для диагностики первого приступа А. при ПБ [1, 4]. Желчный пузырь, как правило, несколько увеличен, застойный и находится в нежных спайках с окружающими тканями или органами, в основном с сальником. В серозной жидкости, взятой из брюшной полости, у всех больных обнаруживаются в различных количествах лейкоциты и лимфоциты. Окончание приступа сопровождается обильным потоотделением на фоне снижения t° тела, началом отхождения газов, уменьшением А. У некоторых больных A. сочетаются с торакалгиями колющего или давящего характера, затрудняющими дыхание, и суставными явлениями. При выраженных А. нередко производятся неоднократные лапоротомии, «ненужные операции» — аппендэктомии, холецистэктомии, спленэктомии, грыжесечение, нефрэктомии, гистеровариэктомии. У таких пациентов описан феномен «географического живота», характеризующегося наличием многочисленных послеоперационных рубцов [1, 4, 6, 8, 9].
Указанные симптомы возникают с интервалом несколько недель или месяцев и обычно проявляются в течение 1–3 дней. Пароксизмы рецидивируют с частотой от 1-2 нед до 1 года; могут быть спонтанные ремиссии. Редко наблюдается status periodicus, когда приступы повторяются практически без светлых промежутков; очень редко возможны и абортивные приступы. Имеются наблюдения развития периодической пурпуры, язвенного поражения кожи лица, нейтропении, менингита, психоза, эпилептиформных судорог [4, 6, 9]. Ассоциированный с ПБ; вторичный АА-амилоидоз развивается, по одним данным, в 25%, по другим — в 41,3% случаев, особенно у носителей HLA A28, B5. Поражение почек часто определяет прогноз (причина смерти больных от уремии до 40-летнего возраста) и считается наиболее постоянным и выраженным признаком при ПБ с лихорадкой, абдоминальными и плевральными синдромами. Известны 2 фенотипа ПБ: при первом (чаще) — присоединение амилоидоза происходит при уже имеющейся картине ПБ; при втором (реже) — амилоидоз является первым признаком заболевания. Наряду с этим встречаются случаи ПБ без амилоидоза и случаи, когда амилоидоз служит единственным проявлением заболевания [8].
Описанный клинический случай представляет интерес для врачей различных специальностей с целью проведения дифференциального диагноза НАО и ПБ.
Впервые приступообразные боли в животе (абдоминалгии) появились в возрасте 7 лет. А. продолжительностью более 12 ч повторялись каждые 7–12 дней, сопровождались рвотой, жидким стулом, при этом не отмечалось связи с конкретными причинными факторами, провоцирующими приступы. Неоднократное обследование у различных специалистов, патологии не было выявлено, еженедельные приступы сохранялись. В 13 лет на фоне очередного приступа абдоминалгии госпитализирована в хирургическое отделение с подозрением на холецистит, при лапаротомии изменений желчного пузыря не выявлено, произведена аппендэктомия. Посев на флору серозной жидкости подтвердил асептическое воспаление. С 15 лет на фоне месячных отмечается появление рецидивирующих спонтанных О. различной локализации кожи и слизистых оболочек, исчезающие самостоятельно через 12–16 ч, возникающие еженедельно. О. на коже и слизистых возникали с той же периодичностью, как и приступы А., иногда провоцировались травмой, психоэмоциональной нагрузкой, стоматологическими манипуляциями (экстракция зуба). В 1974 г. впервые О. гортани с удушьем. Применение антигистаминных препаратов и глюкокортикостероидов без положительного эффекта. Со слов пациентки и по данным медицинской документации, лихорадочных реакций, кратковременного повышения температуры тела до, во время и после приступов А. никогда не отмечалось (кроме редких случаев ОРВИ с t до 37,5°С). Больная неоднократно обследовалась у аллерголога, данных, свидетельствующих о наличии аллергопатологии, получено не было, протеинурии не отмечалось. Во время беременности прослеживалась транзиторная протеинурия до 0,03 г/л/сут, умеренное повышение АД до 150 и 90 мм рт. ст., приступы А. продолжались. Роды протекали без развития О. Но после родов О. продолжали возникать через 10–14 дней с той же периодичностью. После родов протеинурия не обнаружена при многократных повторных анализах мочи. После наступления менопаузы абдоминальные боли стали беспокоить реже (1-2 раза в месяц), О. гортани 1-2 раза в год, последний раз в феврале 2001 г.
С марта 2001 г. наблюдалась в Клинике терапии, нефрологии и профессиональных болезней им. Е. М. Тареева ММА им. И. М. Сеченова. Проводилисьобщеклиническое обследование, биопсия десны и прямой кишки. Амилоидоз не верифицирован. Методом исключения, учитывая анамнез, национальность, характерные клинические проявления, невыявленную острую хирургическую патологию при повторяющихся пароксизмах А., установлен диагноз: «периодическая болезнь с абдоминалгиями, ангионевротическими отеками». Назначена терапия колхицином по 1,0 мг/сут в течение года, затем в связи с недостаточным эффектом терапии колхицин назначен по 1,5 мг/сут в течение 6 мес, положительного эффекта от терапии не отмечалось, приступы А. и О. продолжались с той же частотой. В октябре 2002 г. пациентка обратилась в Центр медицинской генетики НАН (г. Ереван). Обнаружена мутация М694V в гетерозиготном состоянии (ген MEFV). Диагноз ПБ не подтвержден. Система комплемента специально не исследовалась. По данным медицинской документации Клиники им. Е. М. Тареева, в связи с № АД в 2001 г. рекомендован прием энапа, но, со слов пациентки, препарат она не принимала.
При поступлении состояние относительно удовлетворительное, t — 36,8°C Конституция гиперстеническая (масса тела — 84 кг, рост — 164 см, ИМТ — 31,23). Кожные покровы бледно-розовые, с несколько повышенной сухостью и нормальной эластичностью, зуда и О. нет. Периферические лимфатические узлы не увеличены. Движения в суставах сохранены в полном объеме, крепитации, болезненности не отмечается, мышцы и скелет без особенностей. ЧДД 18/мин, дыхание везикулярное, хрипов нет. Тоны сердца ясные с ЧСС = 84/мин, ритм правильный; дующий систолический шум на верхушке. АД 145 и 90 мм рт. ст. При осмотре О. слизистой оболочки полости рта нет. Живот увеличен в размерах, симметричный, мягкий, б/б при поверхностной пальпации. При глубокой ориентировочной методической скользящей пальпации по Образцову-Стражеско-Василенко симптомов раздражения брюшины не выявлено. Слабоположительные симптомы: Ортнера и Георгиевского-Мюсси справа. Печень и селезенка не увеличены. Стул и мочеиспускание регулярные, свободные, безболезненные, симптом «поколачивания» отрицательный с обеих сторон. При исследовании поджелудочной железы: дискомфорт при пальпации зоны Шоффара и панкреатической точки Дежардена. Щитовидная железа мягко-эластической консистенции, не увеличена.
При копрологическом обследовании выявлены признаки ферментативной недостаточности поджелудочной железы. При биохимическом анализе крови: снижение уровня сывороточного железа до нижней границы нормы при ОЖСС — 46,2, остальные основные показатели в пределах нормы. Во время умеренного приступа абдоминальных болей: АЛТ 18,2 ед/л, АСТ 21,0 ед/л, ЩФ 65 ед/л, a-амилаза 39 ед/л, диастаза мочи 110 ед/л. Выявлено снижение g-глобулинов до 7,0 г/л (в норме 8,3–16,8 г/л). Коагулограмма (вне приступа): признаки гиперкоагуляции. При УЗИ: диффузные изменения печени и поджелудочной железы по типу липоматоза; в остальном без патологии. ЭКГ: ритм синусовый, правильный с ЧСС 68/мин. Полугоризонтальное положение ЭОС. Замедление АВ-проводимости I степени. Изменения миокарда диффузного характера. В иммунограмме: CD 8+абс. — 386 (нижняя граница нормы), CD4+/CD8+ — 2,52 (N 1,0–2,5), CD 19+ — 3% (N 5-19), CD 19+абс. — 61 (N 100-500), ЦИК (у. е.): ПЭГ 3% — 32 (N 14-35), ПЭГ 4% — 96 (N 50-90), соотношение 3% к 4% — 3,0 (N 2,5-3,5), показатели Ig A, M, G, общего E в норме. Скарификационные кожные аллергопробы: тест-контроль — «-», гистамин — «++». Сенсибилизации к бытовым, эпидермальным и пыльцевым аллергенам не выявлено. При исследовании системы комплемента в Институте иммунологии МЗ РФ: С2 — 2 (4-6), С4 — 2 (5-7), СН50 — 3 (4-6), С1inh — 30 ед (в 5 раз ниже минимально нормального значения). Показатели снижены. С2 — 2 (4-7), С4 — 6 (7-10), С5- 5 (5-8), СН50- 5 (4-7). Заключение: С2, С4 — ниже нормы, С5 — нижний предел нормы. Очень низкая активность комплемента. Острофазное состояние. При обследовании также получено сомнительное количество антител к антигенам лямблий. При копроовоцистоскопии (ФЦ Госсанэпиднадзора) — цисты Lamblia intestinalis. В зеве: Str. salivarius — умеренный рост, Candida albicans — обильный рост.
Учитывая данные анамнеза заболевания, характер клинической картины А., появление АНО без крапивницы, течение заболевания с детского возраста, отсутствие протеинурии, лихорадки и амилоидоза, неэффективность антигистаминных и глюкокортикостероидных препаратов, отрицательный эффект от терапии колхицином и выраженный положительный эффект при назначении Э-АКК на фоне резкого ѓ фракций комплемента — С4, С2, С1inh поставлен следующий диагноз: наследственный ангионевротический отек с количественным дефицитом С1-ингибитора, пароксизмальные абдоминалгии, стадия медикаментозной ремиссии при выписке. Хронический гастродуоденит, вне обострения. Хронический холецистит, вне обострения. Дискенезия желчевыводящих путей. Жировой гепатоз. Хронический панкреатит. Хронический колит. Лямблиоз кишечника. Артериальная гипертензия II степени. Хроническая венозная недостаточность 0-I ст. Ожирение II степени. Остеохондроз поясничного отдела позвоночника с корешковым синдромом, вне обострения. Вегетативно-сосудистая дистония с явлениями церебральной вазопатии. Ангиодистония сетчатки. Пациентке выдан паспорт, подтверждающий диагноз НАО. Проведена противопротозойная терапия (макмирор, тинидазол), полоскание горла антисептиками, энзимотерапия (мезим-форте), назначена гипотензивная терапия арифоном. За время пребывания в стационаре больная отмечала приступ болей в правом подреберье, чувство дискомфорта в грудной клетке, было проведено обследование, данных, свидетельствующих о наличии острого панкреатита, холецистита на УЗИ, не получено, приступ купирован приемом анальгетиков, в/в введением Э-АКК. Начата базисная терапия дановалом в суточной дозе 600 мг. В дальнейшем состояние больной стабилизировалось, самочувствие улучшилось. О. слизистых и кожи нет. Дыхание и гемодинамика в норме. Рекомендовано продолжить прием дановала 600 мг/сут и завершить коррекцию выявленных нарушений, а также обследование всех родственников. Пациентка более 12 мес находится на терапии дановалом в дозировке сначала 600 мг/сут, далее через 3 мес по 200 мг/сут. О. кожи и слизистых не отмечается. Рекомендации по лечению и ведению пациентов при НАО [2–3, 10–12, 14]
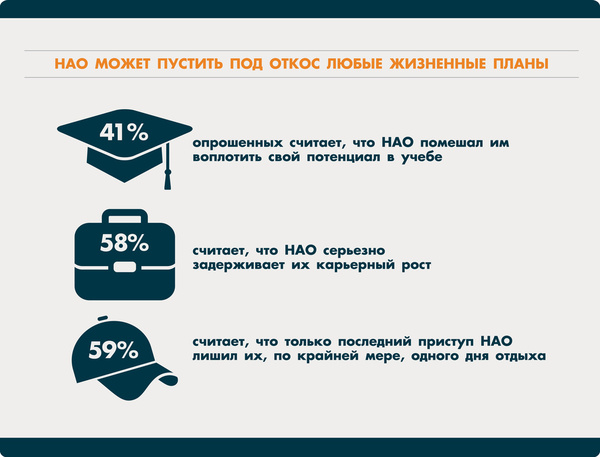
Пациенты с НАО требуют не только дифференцированного индивидуального подхода, но и назначения препаратов, учитывая возможный риск, изменение качества жизни. Такие предпосылки обусловлены тем, что дебют заболевания чаще происходит в пубертатный период, когда имеются не только эндокринные, но и психологические проблемы у пациентов с НАО.
В случае дебюта НАО у девочек в пубертатном периоде или женщин во время беременности и лактации рекомендуется начинать лечение с введения нативной плазмы. Обязательный прием даназола (дановала) постоянно, по жизненным показаниям, ежедневно по схеме: 200 мг 3 раза в день (600 мг) в течение 10–14 дней, затем 200 мг в день (по 1 табл. 2 раза в сутки) 1 мес, 100 мг/сут 6 мес, изменение дозы возможно под контролем аллерголога. Или прием метилтестостерона в начальной дозировке 0,01 г (по 0,005 г 2 раза в сутки, сублингвально; максимальная суточная доза составляет не более 0,025 г) ежедневно. Контроль в крови комплемента для коррекции дозы даназола.В период обострения НАО, при отеке жизненно важных органов: свежезамороженная (или свежая) нативная плазма не менее 250–300 мл в/в одномоментно, капельно (хранение плазмы при -30°С возможно не более 2 мес) или 5% р-р Э-АКК по 100–200 мл в/в капельно, затем по 100 мл в/в капельно каждые 4 ч или 4 г Э-АКК внутрь до полного купирования обострения (4-5 раз в день). Для купирования острых О. введение Э-АКК менее эффективно, чем нативной плазмы.Вместо Э-АКК можно использовать транексамовую кислоту 1-1,5 г внутрь 2-3 раза в день.Для лечения острых приступов применяют очищенный концентрат С1inh 3000 — 6000 ЕД 1-2 ампулы. Рекомендуется применять очищенный С1inh не менее 3 раз в год. К сожалению, препарат С1inh дорогостоящий и не зарегистрирован в РФ, поэтому не может рекомендоваться для широкого использования в нашей стране.При О. в области лица и шеи показано в/в капельное введение свежезамороженной нативной плазмы 250–300 мл, Э-АКК 5% 200–300 мл, лазикс 40–80 мг, дексон 8–12 мг в/в.При отеке гортани ингаляционно 0,1% р-р адреналина, 5% р-р эфедрина, В-адреностимуляторы, госпитализация в ЛОР-отделение.При развитии абдоминального синдрома — наблюдение хирурга. При выраженных болях, рвоте, поносе необходимо введение анальгетиков и спазмолитиков (баралгин, платифиллин, но-шпа и др.) под постоянным наблюдением хирурга, так как длительно существующий О. стенки кишечника может вызвать некроз и потребовать оперативного лечения.Больным с относительными противопоказаниями к приему даназола и метилтестостерона прием Э-АКК 4–12 г/сут под контролем свертывающей системы крови каждые 10–14 дней.Перед срочным оперативным вмешательством показано в/в капельное введение 250–300 мл свежезамороженной нативной плазмы, 200–300 мл 5% Э-АКК, 8–12 мг дексазона (90–120 мг преднизолона).Перед плановым оперативным вмешательством: Э-АКК по 4 г 4 раза в сутки в течение 2 дней до и 2 дней после операции; при невозможности перорального приема Э-АКК вводить ее в/в в количестве 200,0 мл 5% р-ра перед операцией и 4-5 раз по 100,0 мл в течение первых суток после операции; перед операцией ввести 250–300 мл свежезамороженной нативной плазмы.Следует избегать нахождения и работ в сырых, запыленных помещениях, психоэмоциональных нагрузок. Противопоказаны интенсивная физическая нагрузка, механическое воздействие на кожу и слизистые оболочки. Своевременная санация очагов хронической инфекции. Лечение антибиотиками строго по показаниям и по назначению врача, с определением чувствительности микрофлоры, при необходимости, применение противогрибковых средств.Исключить препараты пенициллинового ряда и их производные, В-блокаторы, фитопрепараты, ингибиторы АПФ.Динамическое наблюдение аллерголога-иммунолога, других специалистов по показаниям.Пациенты с НАО нередко нуждаются в психологической и социальной реабилитации. Для многих пациентов зрелого возраста характерно многолетнее и безуспешное лечение у врачей различных специальностей — терапевтов, гастроэнтерологов, гинекологов, хирургов, врачей приемных комиссий военкоматов и др. В связи с этим больным НАО часто ставят неправильный диагноз (О. Квинке, крапивница, аллергия, острый живот, острый аппендицит, острый холецистит, стеноз чревного ствола, ангина, ревматологическое заболевание, кровоизлияние в мозг, мигрень, эпилепсия и др.) и назначают неадекватную для данного заболевания терапию, что становится причиной высокой смертности таких пациентов. Около 25% больных умирают от О. гортани в возрасте до 30 лет. Многим больным неоднократно проводят лапаротомии и трахеотомии из-за некупирующихся О. [2, 3, 10, 13].
Данный клинический случай ярко демонстрирует сложности дифференциальной диагностики абдоминального синдрома при НАО и ПБ. Это особенно важно для практикующих врачей-хирургов, терапевтов, врачей скорой помощи, оториноларингологов, аллергологов. От клиницистов требуется не только сбор анамнеза заболевания и владение методами объективного исследования, но и знание клинической картины кожно-висцерального синдрома — ПБ и атипичных проявлений НАО, ставших актуальной проблемой современной клинической аллергологии и иммунологии.
Г. Х. Викулов Е. С. Феденко, доктор медицинских наук Т. В. Латышева, доктор медицинских наук, профессор ГНЦ Институт иммунологии МЗ РФ, г. Москва
Примечание. Статья взята с сайта http://www.lvrach.ru. Оригинал статьи доступен по адресу http://www.lvrach.ru/2004/03/4531137/